案例:先天性髓鞘發育不良神經病
臨床信息:男,7天,反應差,不哭,不能吸吮,吞咽差,雙足內翻,肌張力低。2020/06/10出現抽搐。超聲:卵圓孔未閉,三尖瓣輕度反流。診斷:1.新生兒肺炎,2.早產兒,3.新生兒呼吸窘迫綜合征?,4.新生兒窒息。
檢測項目及內容:全外顯子組測序;本全外顯子組基因檢測針對人類基因組的外顯子組的全部區域,覆蓋20000多個基因,涵蓋85%以上的人類遺傳性疾病。檢測范圍包括基因編碼區單核苷酸位點變異(SNV)、小片段插入/缺失(INDEL)等突變類型。
檢測結果:檢出2個匹配受檢者臨床表型的可能致病性的基因變異:
CNTNAP1基因c.2590_2593del:p.H864Qfs*20雜合變異
CNTNAP1基因c.C1294T:p.Q432X雜合變異
基因變異位點:
CNTNAP1基因關聯一組常染色體隱性遺傳的神經病,OMIM收錄臨床表型為:

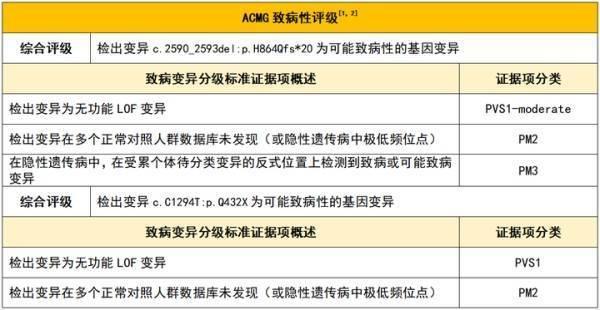
ACMG致病性評級:
一代驗證測序結果:
附:一代測序圖
CNTNAP1:NM_003632:exon17:c.2590_2593del:p.H864Qfs*20
先證者該位點為雜合型先證者父親該位點為雜合型先證者母親該位點為野生型
CNTNAP1:NM_003632:exon8:c.C1294T:p.Q432X
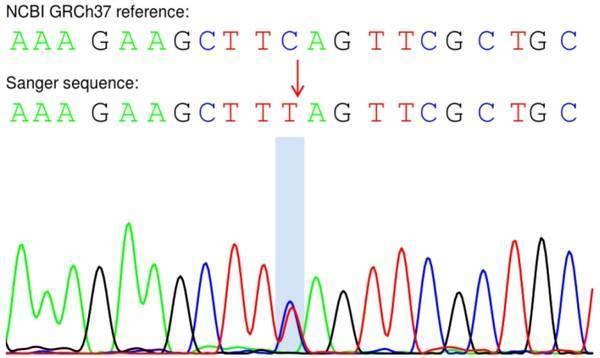 先證者該位點為雜合型先證者父親該位點為野生型先證者母親該位點為雜合型
先證者該位點為雜合型先證者父親該位點為野生型先證者母親該位點為雜合型
遺傳咨詢建議
1. 受檢者將來婚育,其下一代有100%概率遺傳到上述兩個變異之一,為攜帶者。建議受檢者將來配偶行遺傳咨詢和基因檢測。
2. 受檢者父母均為雜合子攜帶者,在常染色體隱性遺傳模式下,雜合子攜帶者一般不表現疾病病理表型。受檢者父母若再生育,子代有25%的概率同時遺傳到上述兩個變異。建議生育前再行遺傳咨詢。
先天性髓鞘發育不良神經病
先天性髓鞘發育不良神經病(Congenital hypomyelination neuropathy,CHN)是一種出生時出現的神經系統疾病。主要癥狀包括呼吸困難、肌肉無力和不協調、肌張力差(新生兒張力減退)、反射消失(無反射)、行走困難(共濟失調),和/或感覺或移動身體某部分的能力受損。先天性髓鞘發育不良神經病3型(CHN3)是其中一種,CHN3是一種嚴重的常染色體隱性遺傳神經病,是由染色體17q21上CNTNAP1基因的純合或復合雜合突變引起的,其特征是子宮內神經源性肌肉損傷,由於軸突髓鞘化不當而導致的早期張力減退、無反射、遠端肌肉無力和非常慢的神經傳導速度(NCV)。在其極端形式,它可能表現為嚴重的關節攣縮或先天性多發性關節攣縮癥和呼吸功能不全。在不太嚴重的情況下,患者可能可以步行。先天性髓鞘發育不良神經病的癥狀和這些癥狀的嚴重程度因人而異。受累個體的精神運動發育嚴重受損,可能在嬰兒期或幼兒期死亡。
臨床特征
CNTNAP1(MIM#602346)編碼CASPR,並於2014年由 Laquerriere等人首次研究與人類疾病有關。Low, K., Stals, K.等人於2018年研究瞭來自6個不同傢庭的3名女性和4名男性的臨床和分子數據,7名患者都有嚴重的智力障礙(ID)和嚴重的神經表型。沒有一個病人有觀察到其他基因組的異常[3]。
圍產期:除一例妊娠外,所有妊娠均出現明顯羊水過多。這與5例早產有關,但沒有患者在妊娠30周以下出生。所有患者的出生體重和頭圍均在正常范圍內,但其中6例的呼吸功能極低,重度肌張力減退,需要快速復蘇。患者7是受影響最重的,在3個月大時死亡。
神經病學、神經病理學與發育:所有病例均有中樞性和外周性張力減退,伴有不同程度的脫髓鞘/髓鞘發育不良神經病變和各種可變的附加神經癥狀:1例患者有眼球震顫、共濟失調、震顫等,2例有肌張力障礙。MRI腦掃描一致顯示髓鞘發育不良/脫髓鞘,白質體積不同程度減少和腦萎縮。患者1、2、3、4的神經活檢結果如圖1所示。
圖1
圖1 A,B.正常神經,橫切面,(A)H&E 40 ×, (B)Sol. Cyan 40 ×;C,D.正常神經,縱切面,(C)H&E 40 × ,(D)Sol. Cyan 40 ×;所有正常的神經切片都顯示瞭適當厚度的有髓神經纖維的正常數量;E,F.患者1:9歲時腓腸神經活檢(TS)在H&E (E)和 Sol. Cyan (F)上顯示大直徑(厚髓鞘)神經纖維明顯缺失,細的有髓纖維叢生(軸突出芽),與髓鞘發育不良神經病一致;G,H.患者2:10個月時腓腸神經活檢(TS)在H&E (G)和 Sol. Cyan (H)上顯示厚髓鞘的適度丟失,出現細的髓鞘纖維,有細的髓鞘纖維簇和罕見的洋蔥鱗莖;I,J.患者7:3個月大死亡後的腓腸神經活檢(屍檢標本)在H&E (I)和 Sol. Cyan (J)上顯示廣泛的幾乎所有的有髓纖維丟失伴有殘留的細髓鞘纖維;K.患者2:電子顯微術(EM)突出瞭細的髓鞘纖維簇,插圖突出瞭單個洋蔥鱗莖;I.患者7:EM顯示廣泛有髓纖維丟失伴有殘留的細髓鞘纖維;M.正常EM。
呼吸系統:7例患者中有6例在出生時出現嚴重呼吸窘迫,5例需要氣管切開。
畸形:病人的面部特征如圖2所示。所有患者都有一致的肌病性面部特征和鮮紅斑痣(葡萄酒樣痣、葡萄酒色斑)。
圖2
圖2 患者1、2、3、4、6、7面部特征從左至右。患者1的表型為最輕度的。註意到有狹窄的下斜瞼裂,豐滿的眉毛,肌病性面容和張大的嘴巴。患者2、3、4、6均行氣管造口術。
其他特征:牙齦增生2例,其中1例有額外牙齒。所有的患者都有典型的“教堂樣”高腭穹,但隻有患者7有腭裂。患者5有前喉的喉骨軟化。患者6有不明原因的靜息性心動過速。患者4有小指甲,嚴重濕疹,皮膚色素沉著異常。
鑒別診斷
CNTNAP1基因(接觸蛋白相關蛋白1)(OMIM:602346)突變與致死型先天性攣縮綜合癥7型(LCCS7)(OMIM:616286)和先天性髓鞘發育不良神經病3型(OMIM:618186)這兩種罕見的常染色體隱性遺傳性先天性疾病有關。CNTNAP1基因中的純合或復合雜合突變可能導致CHN3,但LCCS7僅由純合突變引起[6]。LCCS7是先天性多發性關節攣縮癥(AMC)的一種軸突膠質形式,其特征是先天性遠端關節攣縮、羊水過多、胎動減少和嚴重的運動麻痹,導致新生兒早期死亡。
先天性多發性關節攣縮癥(AMC)與胎兒運動減退和潛在的神經系統疾病有關,它可能是由於骨骼肌,或神經肌肉連接或軸突膠質的發育受損。然而,也可以由非遺傳因素引起,即母親自身免疫性肌無力或胎動受限。根據Hengel等人在2017年的記錄,AMC不再是髓鞘發育不良神經病的一個特征,可能是肌肉骨骼疾病的主要表現,也可能是神經系統進程的次要表現。因此,與LCCS7相反,CHN3與AMC無關[6]。
CHN最初由Dejerine和Sottas在19世紀晚期提出,主要由漸進性肌肉萎縮組成,後來出現瞭新的分類。CHN表現為肌張力減退、反射減退和遠端肌無力。CHN1(OMIM:605253)通常與EGR2基因突變有關,而CHN2(OMIM:618184)則與MPZ基因突變有關。有趣的是,與CHN3相反,在這兩種類型中沒有智力殘疾的報告。神經組織學在CHN伴有嚴重髓鞘發育不良的病例中仍然是非特異性的,而其他髓鞘發育不良的疾病,如Dejerine-Sottas綜合征或Charcot-Marie-Toth 1型,則發現有多餘的“洋蔥鱗莖”形成[6]。
Dejerine-Sottas病是一種遺傳性神經系統疾病,可逐漸影響肌肉功能。周圍神經變大變粗,導致肌肉無力的不規則發展。疼痛、無力、麻木和刺麻感、針刺痛或灼熱感可能發生在患者的腿上。其他癥狀包括熱敏感度喪失、反射消失和腿部肌肉萎縮。手和前臂的肌肉在後期可能會變得無力。也可能出現輕度視力障礙[4]。
格林-巴利綜合征(急性特發性多神經炎)發生在人體免疫系統(抗體、淋巴細胞)攻擊神經,損害神經髓鞘和軸突時。神經信號被延遲和改變,導致腿部、手臂和身體其他部位的肌肉無力和癱瘓,並伴有感覺異常。當肌肉神經受損時,患者會感到肌肉酸痛和無力,難以從椅子站起來或走樓梯,難以舉起物體,呼吸急促和/或吞咽困難[4]。
參考文獻:
[1]Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
[2]王秋菊, 沈亦平, 鄔玲仟,等. 遺傳變異分類標準與指南[J]. 中國科學:生命科學, 2017(6).
[3]Low K J, Stals K, Caswell R, et al. Phenotype of CNTNAP1: a study of patients demonstrating a specific severe congenital hypomyelinating neuropathy with survival beyond infancy[J]. European Journal of Human Genetics, 2018, 26(6): 796-807.
[4]https://rarediseases.org/rare-diseases/neuropathy-congenital-hypomyelination/
[5]https://www.uniprot.org/uniprot/P78357
Sabbagh S, Antoun S, Mégarbané A. CNTNAP1 Mutations and Their Clinical Presentations: New Case Report and Systematic Review[J]. Case Reports in Medicine, 2020, 2020.