代謝物組經過一系列專業檢測後,應該會得到一個類似於基因表達的矩陣,接著就這矩陣數據進行分析。根據檢測方法不同,前期數據的處理方法可能略有不同,本文選用absoluteIDQ p180 kit ( mass spectrometry質譜原理)產出數據進行數據分析。
分析流程
一般在遞交樣本分析時,可能會一同提交幾份均勻混合瞭全部樣本的QC樣本(一起混合再分成幾份)。以此來檢測檢測過程中的穩定性和做後期質量控制。QC樣本結果需要相對集中(PCA時)證明全程分析穩定。QC樣本的數值幫忙做樣本內normalization,保證樣本間數據有可比較性。
分析方法:

在線分析軟件 MetaboAnalysis 4.0
Chong, J., Wishart, D. S., & Xia, J. (2019). Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Current Protocols in Bioinformatics, 68(1), 1–128. https://doi.org/10.1002/cpbi.86
中文教程:
這個軟件和文章基本上都能滿足代謝物的簡單分析,論文中有很詳細的官方使用步驟&參數說明。這款軟件基於R來完成,最終結果和突變可以直接在網上下載下來,也挺方便的。在線版軟件在執行過程中可以顯示對應的R命令行,也有配套的R包,可以自己下載下來學習使用。由於是在線版本,參數選擇和結果顯示內容都不是太細節,有一寫數據無法拿到,所以接下來說說自己用代碼分析的操作。
代碼分析步驟
一、Sample Quality Check
配合QCsample來檢查前期實驗檢測的穩定性和可靠性。一般千萬要理解好數據再開始分析,不然最終結果的可信度和真實性也無法保障。瞭解檢測機制,可能引起的檢測誤差,樣本間的濃度處理等,在此檢查樣本分組有無出錯等。本次使用的數據物QC sample,掠過。可以參考資料:https://www.futurelearn.com/courses/metabolomics/0/steps/10703
二、數據預處理
本文的數據預處理方式參考選用MetaboAnalysis4.0 的處理方式。下文代謝物的不同種類稱為不同feature。基本數據預處理方法如下,當然針對不同的數據可能還會有其他分析步驟,如分組/集中之類的,由於我數據分析過程中未執行,此處不詳述。
- 過濾低質量feature。一般一種代謝物超過50%檢測不到(含量太低導致的數據缺失),去除這個feature,不參與後續分析。我的數據有80個人,188種代謝物,所以限制得更為嚴格,需要70個以上樣本有檢出結果。
- 缺失數據處理impute missing data。 代謝物檢測一般有最低檢測數值(LOD),意思是檢測樣本中該代謝物含量必須要達到某個值濃度才可以被檢測出來。在這裡處理missing data 的方式就是用0.5* LOD的方式補充缺失數據。
- scalling :把數據壓縮到一個范圍內。在這裡直接選用MetaboAnalysis4.0推薦的auto scalling模式。mean-centered and divided by the standard deviation of each variable。
- 數據標準化normalization, 在樣本間做數據標準化,這一點非常重要。一般標準化後的數據可以得到正態分佈的數據集。有不同的數據標準化方式,分析的時候我前後選用瞭 normalization by Sum (MetaboAnalysis 推薦方式), noramlization by Median, normalization by quantiles 方法,前兩個標準化方法最終出來的分析結果都不大好(數據分得不明顯),所以一直嘗試到四分數標準化方法。具體數據具體分析,大傢可以再多研究。關於標準化方法:
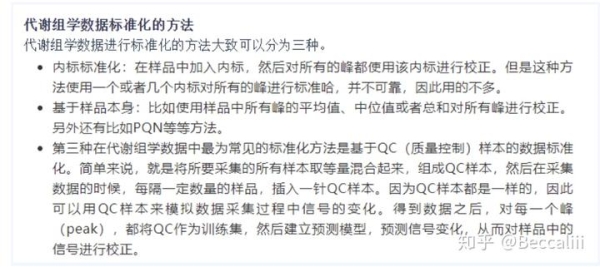
針對代謝組的數據標準化方法介紹,還有一篇文講得蠻好的,這位作者還有其他的關於代謝組的文章,也非常有幫助,感謝分享:
經過標準化後的樣本數據:
 右邊的“bell shape”圖是我們想看到的結果
右邊的“bell shape”圖是我們想看到的結果
關於數據分析 一般會先看一下樣本分佈
– 每個樣本的總濃度,正態分佈是我們想看到的,差別過大是我們需要註意到。
– 簡單看一下case和control組的差別,我們希望兩組數據應給沒有太大的差別,因為先驗知識告訴我們代謝物可能並不會在總體分佈上在case和control組有太大差別。所以如果兩組數據有明顯差別,有可能是兩組樣本在實驗檢測時出瞭差別。所以需要樣本間做normalization來消除誤差
– 某些feature的數值是否占比太大,如在noramlization by sum的方法,基本原理是把絕對值濃度轉換成樣本中占比來計算。但feature有一個H1濃度明顯整體偏大,由此使其他樣本但占比更小,太小但數據件差別就被模糊瞭,所以效果不好。
可以先觀察一下數據分佈特征來做出最合適的數據預處理。
包括前期數據預處理,樣本數據分析和feature數據分析(是否需要去除outlier)之類的,數據下載。處理數據部分由代碼完成,數據預處理完成後下載數據,並輸入MetaboAnalysis在線版本進行畫圖(PCA,火山圖,熱圖,RF)之類。 當然也可直接用R完成,代碼見下文。
三、數據分析
- PCA 檢查:QC是否聚集,是否樣本天然分組符合預期,離群樣本/異常樣本等
- 熱圖 和PCA類似,相互驗證,可以移除outlier後,讓case和normal組能自然聚合在一起。
- 火山圖 得到差異表達的代謝物,一般高表達和低表達的都看一下
- PLS-DA 針對數據,建立預測模型,並查看模型的預測正確率。查看錯分類的樣本,看是否能調整,提高模型預測準確度(Q2越接近1越好)。
PLS-DA 分析,對訓練集和測試集的性能測試
- 通路分析 得到一系列差異表達的代謝物後,可以直接借用MetaboAnalysis的通路分析,來看哪些代謝活動最受影響。
後續分析 R代碼實現
PCA
data_pca<-as.data.frame(read.table("data/IDQ180_filter_impute_qnor_JCsample2.csv",sep=",",header=T,row.names=1,stringsAsFactors=F))
data_pca=t(data_pca)
#convert to numeric label
table(data_pca[,1])
a <- sub("F",2,data_pca[,1])
b <- sub("M",1,a)
data_pca[,1] <- a
data_pca[,1] <- b
data_pca<-as.data.frame(data_pca)
data_pca[1:2,]
##convert to numeric !really helpful
data_pca[] <- lapply(data_pca, function(x) {
if(is.factor(x)) as.numeric(as.character(x)) else x
})
data_pca[1:2,]
dim(data_pca)
# apply PCA - scale. = TRUE is highly advisable, but default is FALSE.
out_pca <- prcomp(data_pca[,-1])
#out_pca[2]
write.csv(out_pca[2],file="data/qnorm_34JCsamples_pca.txt",quote=F,row.names=T)
plot(out_pca,type="l")
#install.packages('ggfortify')
library(ggfortify)
autoplot(prcomp(data_pca[,-1]),data=data_pca,colour='sex',size=1,label=TRUE,label.size=3)
#remove sample JC50023PL.02 and redo PCA plot
data_pca<-data_pca[!rownames(data_pca) %in% c("JC50023PL.02"),]
autoplot(prcomp(data_pca[,-1]),data=data_pca,colour='sex',size=1,label=TRUE,label.size=3)